Efavirenz
Princípio/forma ativa - Bula - Efavirenz
Para que serve?
Efavirenz é indicado para o tratamento antiviral combinado de adultos, adolescentes e crianças infectados pelo HIV-1.
Contraindicação
Efavirenz é contraindicado para pacientes com hipersensibilidade clinicamente significativa a qualquer um de seus componentes.
Efavirenz não deve ser administrado concomitantemente com as doses padrões de voriconazol uma vez que o efavirenz reduz significativamente as concentrações plasmáticas do voriconazol ao passo que o voriconazol também aumenta significativamente as concentrações plasmáticas do efavirenz.
Pacientes utilizando efavirenz não devem usar concomitantemente produtos contendo Erva-de-são-joão ( Hypericum perforatum ) uma vez que pode ser esperada redução das concentrações plasmáticas de efavirenz. Este efeito é devido à indução da CYP3A4 e pode resultar em perda de eficácia terapêutica e desenvolvimento de resistência.
Posologia e como usar
Uso oral.
A dose de Efavirenz solução oral deve ser medida utilizando-se a seringa para administração oral inserida na embalagem.






Após o uso, mergulhe a seringa em água quente com sabão durante pelo menos um minuto. Depois, aspire a água, enchendo a seringa completamente e, em seguida, esvazie-a pressionando o êmbolo até o fim. Repita este procedimento três vezes, no mínimo. Separe o êmbolo do corpo da seringa e lave bem as duas partes em água corrente quente. Se alguma parte da seringa não ficar limpa, repita o procedimento.
Deixe secar completamente as partes separadas da seringa antes de montar novamente o conjunto. Não lave a seringa na máquina de lavar pratos.
Posologia do Efavirenz
Efavirenz pode ser tomado com ou sem alimentos, conforme desejado.
A posologia recomendada de Efavirenz em combinação com um inibidor de protease e/ou ITRNs é de 600 mg, por via oral, uma vez ao dia.
A fim de melhorar a tolerabilidade às reações adversas neurológicas, recomenda-se a administração ao deitar durante as primeiras duas a quatro semanas de tratamento e para aqueles pacientes que continuam a apresentar sintomas.
Efavirenz deve ser administrado em combinação com outras medicações antirretrovirais.
A dose recomendada de Efavirenz em combinação com um inibidor de protease e/ou ITRNs para pacientes com 17 anos de idade ou menos está descrita na Tabela 4. Os comprimidos de Efavirenz devem ser administrados apenas a crianças capazes de degluti-los com segurança. Efavirenz não foi adequadamente estudado em crianças com menos de três anos de idade ou com peso inferior a 13 kg. Efavirenz comprimidos não é adequado para crianças com peso inferior a 40 kg; para esses pacientes, está disponível a apresentação em solução oral.
Tabela 4. Dose Pediátrica a Ser Administrada Uma Vez ao Dia
Efavirenz Comprimidos
Dose (mg)
Efavirenz pode ser ingerido com ou sem alimento, à escolha do paciente.
Reações Adversas
O efavirenz foi geralmente bem tolerado em estudos clínicos que envolveram mais de 9.000 pacientes. Em um subgrupo de 1.008 pacientes que receberam 600 mg ao dia de Efavirenz em combinação com inibidores da protease e/ou ITRNs em estudos clínicos controlados, os efeitos indesejáveis relacionados ao tratamento relatados mais frequentemente, cuja gravidade foi no mínimo moderada, e que ocorreram em pelo menos 5% dos pacientes foram: erupção cutânea (11,6%), tontura (8,5%), náuseas (8,0%), cefaleia (5,7%) e fadiga (5,5%). A frequência dos relatos de náuseas foi mais alta nos grupos controle. Os efeitos indesejáveis associados ao efavirenz que mais se destacam são erupção cutânea, sintomas neurológicos e psiquiátricos. A administração de Efavirenz com alimentos pode aumentar a exposição ao efavirenz e pode levar a um aumento da freqüência de reações adversas.
Outros efeitos indesejáveis relacionados ao tratamento e clinicamente significativos, porém menos frequentes, relatados em todos os estudos clínicos incluem: reações alérgicas, coordenação anormal, ataxia, confusão, estupor, vertigem, vômitos , diarreia , hepatite , diminuição da concentração, insônia , ansiedade , alteração do padrão de sonhos, sonolência, depressão , pensamentos anormais, agitação, amnésia, delírios, labilidade emocional, euforia, alucinações e psicoses.
Outros efeitos indesejáveis, relatados por meio de farmacovigilância, incluem neuroses, reações paranóides, distúrbios de coordenação e equilíbrio cerebelares, convulsões, prurido, dor abdominal, visão turva, rubor, ginecomastia , insuficiência hepática, dermatite fotoalérgica, pancreatite , redistribuição/acúmulo da gordura corporal em áreas como o dorso do pescoço, mamas, abdômen, e retroperitônio, tinido, e tremor.
Poucos relatos de farmacovigilância de insuficiência hepática, incluindo casos em pacientes sem nenhuma doença preexistente ou outros fatores de risco identificáveis foram caracterizados por um curso fulminante, progredindo em alguns casos para transplante ou óbito.
O tipo e a frequência dos efeitos indesejáveis em crianças foram, em geral, semelhantes aos observados em pacientes adultos, à exceção da erupção cutânea, que foi mais frequente e geralmente mais grave nas crianças do que nos adultos.
Em estudos clínicos, 26% dos pacientes tratados com 600 mg de Efavirenz apresentaram erupção cutânea em comparação com 17% dos pacientes nos grupos controle. A erupção cutânea foi considerada relacionada ao tratamento em 18% dos pacientes tratados com Efavirenz. Ocorreu erupção cutânea grave em menos de 1% dos pacientes tratados com Efavirenz e 1,7% descontinuaram o tratamento por causa de erupção cutânea. A incidência de eritema polimorfo ou de síndrome de Stevens-Johnson foi de 0,14%.
Foi relatada erupção cutânea em 58 de 182 crianças (32%) tratadas com o efavirenz em três estudos clínicos por uma mediana de 123 semanas. A erupção cutânea foi grave em 6 pacientes. Antes de iniciar o tratamento com o efavirenz em crianças, deve-se considerar a profilaxia com anti-histamínicos apropriados.
As erupções manifestam-se geralmente como erupções maculopapulares leves a moderadas, que ocorrem nas primeiras duas semanas de tratamento com Efavirenz. Na maioria dos pacientes, as erupções desaparecem no período de um mês com a continuação do tratamento com Efavirenz. Efavirenz pode ser reiniciado em pacientes que interromperam o tratamento por erupção cutânea. O uso de anti-histamínicos e/ou corticosteroides apropriados é recomendado quando Efavirenz for reiniciado.
A experiência com Efavirenz em pacientes que descontinuaram outros agentes antirretrovirais da classe dos ITRNNs é limitada. Dezenove pacientes que descontinuaram a nevirapina em consequência de erupção cutânea foram tratados com Efavirenz; nove desenvolveram erupção cutânea leve a moderada durante o tratamento e dois descontinuaram por causa da erupção cutânea.
Foram relatadas experiências adversas psiquiátricas graves em pacientes tratados com o efavirenz. Em estudos clínicos controlados que envolveram 1.008 pacientes tratados com esquemas contendo efavirenz durante uma média de 1,6 ano e 635 pacientes tratados com esquemas de medicamento-controle durante uma média de 1,3 anos, a frequência dos eventos psiquiátricos graves específicos entre pacientes que receberam efavirenz ou medicamento-controle foram, respectivamente: depressão grave (1,6%, 0,6%), idéias suicidas (0,6%, 0,3%), tentativas de suicídios sem sucesso (0,4%, 0,3%) e reações maníacas (0,1%, 0%). O risco dessas experiências adversas psiquiátricas graves parece ser maior em pacientes com histórico de distúrbios psiquiátricos, nos quais a frequência de cada evento citado varia de 0,3% para reações maníacas a 2% para depressão grave e idéias suicidas.
Sintomas que incluem, mas não se limitam a tontura, insônia, sonolência, dificuldade de concentração e alteração do padrão de sonhos constituem reações adversas relatadas com frequência por pacientes que receberam 600 mg ao dia de Efavirenz em estudos clínicos. Em estudos clínicos controlados, nos quais 600 mg de Efavirenz foram administrados com outros agentes antirretrovirais, 19,4% dos pacientes apresentaram sintomas neurológicos de intensidade moderada a grave em comparação com 9% dos pacientes nos grupos controle. Esses sintomas foram graves em 2,0% dos pacientes que receberam 600 mg ao dia de Efavirenz e em 1,3% dos pacientes nos grupos controle. Em estudos clínicos, 2,1% dos pacientes tratados com 600 mg de Efavirenz descontinuaram o tratamento por causa de sintomas neurológicos.
Os sintomas neurológicos começam geralmente durante o primeiro ou o segundo dia de tratamento e geralmente desaparecem após as primeiras 2-4 semanas. Em um estudo clínico, a prevalência mensal de sintomas neurológicos de gravidade pelo menos moderada entre a 4a e a 48a semana variou de 5% a 9% em pacientes tratados com esquemas contendo efavirenz e de 3% a 5% em pacientes nos grupos controle. Em um estudo com voluntários não infectados, um sintoma neurológico típico começou 1 hora após a dose e durou 3 horas (mediana). A administração ao deitar melhora a tolerabilidade a esses sintomas e é recomendada durante as primeiras semanas de tratamento para aqueles pacientes que continuam a apresentá-los. A redução da dose ou o fracionamento da dose diária não se mostraram benéficos e não são recomendados.
Aumentos de AST (TGO) e ALT (TGP) cinco vezes acima do limite superior da normalidade foram verificados em 3% de 1.008 pacientes tratados com 600 mg de efavirenz; foram observados aumentos semelhantes nos grupos controle. De 156 pacientes soropositivos para hepatite B e/ou C tratados com 600 mg de efavirenz, 7% apresentaram aumento de AST (TGO) cinco vezes acima do limite superior da normalidade e 8%, de ALT (TGP); nos grupos controle (91 pacientes), aumentos dessa magnitude ocorreram em 5% (AST/TGO) e 4% (ALT/TGP) dos pacientes. Foram observados aumentos de gama GT cinco vezes acima do limite superior da normalidade em 4% de todos os pacientes tratados com 600 mg de efavirenz e em 10% dos pacientes soropositivos para hepatite B ou C; nos grupos controle, a incidência de aumentos semelhantes de gama GT foi de 1,5% a 2%, independentemente da sorologia para hepatite B ou C. Aumentos isolados de gama GT em pacientes tratados com efavirenz podem refletir indução enzimática não associada à toxicidade hepática.
Foram observados aumentos do colesterol total de 10% a 20% em alguns voluntários não infectados que receberam efavirenz. Em pacientes tratados com efavirenz + AZT + 3TC, foram observados aumentos em relação aos valores basais do colesterol total sem jejum e do HDL de aproximadamente 20% e 25%, respectivamente; em pacientes tratados com efavirenz + IDV, esses aumentos foram de 40% e 35%, aproximadamente. Os efeitos do efavirenz nos triglicérides e no LDL não foram bem descritos. Em outro estudo, aumentos em relação aos valores basais de colesterol total, HDL-colesterol, LDL-colesterol de jejum, e triglicérides de jejum de 21%, 24%, 18%, e 23%, respectivamente, foram observados em pacientes tratados com efavirenz+ZDV+3TC por 48 semanas. A importância clínica desses achados é desconhecida.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Interação medicamentosa
O efavirenz é um indutor da CYP3A4 e da CYP2B6. Outros compostos que são substratos da CYP3A4 ou da CYP2B6 podem ter suas concentrações plasmáticas reduzidas quando administrados concomitantemente com Efavirenz.
Para orientações sobre a coadministração com fosamprenavir e ritonavir , as informações para prescrição para fosamprenavir cálcico devem ser consultadas.
O efavirenz reduz a exposição ao atazanavir. Consultar as informações para prescrição para orientações sobre a coadministração de atazanavir com efavirenz.
Quando o indinavir em uma dose aumentada (1.000 mg a cada 8 horas) foi administrado com o efavirenz (600 mg uma vez ao dia) a voluntários não-infectados, A AUC e a C vale do indinavir foram reduzidas em cerca de 33-46% e 39-57%, respectivamente, em comparação com quando indivinavir foi administrado isoladamente na dose padrão (800 mg a cada 8 horas). Diferenças similares na AUC e C máx de indinavir também foram observadas em indivíduos infectados pelo HIV que receberam indinavir (1000 mg a cada 8 horas) com efavirenz (600 mg 1x/dia) em comparação com indinavir administrado isoladamente (800 mg a cada 8 horas). A dose ótima de indinavir, quando administrado em combinação com efavirenz, não é conhecida. Aumentar a dose do indinavir para 1.000 mg a cada 8 horas não compensa o aumento do metabolismo do indinavir devido ao efavirenz.
Quando administrado efavirenz 600 mg uma vez ao dia juntamente com indinavir/ritonavir 800/100 mg duas vezes ao dia em pacientes infectados por HIV-1 (n=6), a farmacocinética do indinavir e do efavirenz foram geralmente comparáveis à de voluntários não-infectados.
Uma redução significativa na C mín do lopinavir foi observada quando uma combinação de lopinavir/ritonavir foi coadministrada com efavirenz em comparação ao uso de lopinavir/ritonavir isolado. Deve ser considerado um aumento na dose de lopinavir/ritonavir cápsulas ou solução oral para 533/133 mg (4 cápsulas ou 6,5 mL) duas vezes ao dia, administrada com alimento quando administrada com efavirenz. Consulte a informação sobre prescrição de comprimidos de lopinavir/ritonavir para auxílio na coadministração desta formulação com efavirenz.
Quando Efavirenz (600 mg, uma vez ao dia) é administrado em combinação com darunavir/ritonavir (800/100 mg, uma vez ao dia), pode resultar em C mín subótimo de darunavir. Caso Efavirenz seja administrado em combinação com darunavir/ritonavir, deve-se utilizar o esquema darunavir/ritonavir 600/100 mg duas vezes ao dia. Consulte a informação de prescrição de darunavir/ritonavir para auxílio na coadministração com Efavirenz.
A AUC 12 e a C máx do maraviroc (100 mg 2x/dia) são diminuídas em 45% e 51%, respectivamente, quando administrado com Efavirenz (600 mg 1x/dia) em comparação com o maraviroc administrado isoladamente. Consultar as informações para prescrição para o maraviroc para orientações sobre a coadministração com Efavirenz.
A AUC, C máx e C mín do raltegravir (400 mg, dosé única) diminuíram em 36%, 36% e 21 %, respectivamente, quando administrado com efavirenz (600 mg, 1x/dia) em comparação com raltegravir isoladamente. O mecanismo de interação é a indução da enzima UGT1A1 pelo efavirenz. Não é necessário ajuste de dose para o raltegravir.
Quando Efavirenz (600 mg em dose única ao deitar) e ritonavir (500 mg a cada 12 horas) foram estudados em voluntários não infectados, a combinação não foi bem tolerada e as experiências adversas clínicas (por exemplo, tontura, náuseas, parestesias) e anormalidades laboratoriais (enzimas hepáticas elevadas) foram mais frequentes. Recomenda-se a monitoração das enzimas hepáticas quando Efavirenz for usado em combinação com o ritonavir.
Quando o saquinavir (1.200 mg três vezes ao dia, na formulação em cápsula gelatinosa mole) foi administrado com Efavirenz, a AUC e a C máx do saquinavir foram reduzidas em aproximadamente 62% e 45%-50%, respectivamente. O uso de Efavirenz em combinação com o saquinavir como o único inibidor da protease não é recomendado.
Quando efavirenz (600 mg, uma vez ao dia) foi administrado com boceprevir (800 mg, três vezes ao dia), a concentração plasmática mínima de boceprevir foi reduzida (C mín ↓ 44%). O desfecho clínico da redução observada não foi diretamente avaliado.
A administração concomitante de telaprevir e efavirenz resultou em redução de exposição no estado de equilíbrio para telaprevir e efavirenz. Quando foi administrado telaprevir 1125 mg a cada oito horas com efavirenz 600 mg, 1x/dia, a AUC, C máx e C mín de telaprevir foram reduzidos em 18%, 14% e 25% em relação ao telaprevir 750 mg a cada oito horas administrado isoladamente e a AUC, C máx e C mín de efavirenz foram reduzidos em 18%, 24% e 10%. Consultar as informações para prescrição para telaprevir para orientações sobre a coadministração com Efavirenz.
A rifampicina reduziu 26% e 20%, respectivamente, a AUC e a C máx do efavirenz em 12 voluntários não infectados. A dose de Efavirenz deve ser aumentada para 800 mg/dia quando administrado com a rifampicina em pacientes com peso maior ou igual a 50 kg. Não é recomendado ajuste da dose da rifampicina quando administrada com Efavirenz. Em um estudo que envolveu voluntários não infectados, o efavirenz induziu redução de 32% e 38% da C máx e da AUC da rifabutina , respectivamente, e aumentou a depuração de rifabutina. A rifabutina não exerceu efeito significativo na farmacocinética do efavirenz. Esses dados sugerem que a dose diária de rifabutina deve ser aumentada 50% quando administrada com o efavirenz e que a dose da rifabutina pode ser duplicada para esquemas nos quais a rifabutina é administrada duas ou três vezes por semana em combinação com o efavirenz.
A administração concomitante de 400 mg de Efavirenz uma vez ao dia e 500 mg de claritromicina a cada 12 horas durante sete dias resultou em efeito significativo do efavirenz na farmacocinética da claritromicina. A AUC e a C máx da claritromicina foram reduzidas em 39% e 26%, respectivamente, enquanto a AUC e a C máx do hidroximetabólito da claritromicina foram aumentadas 34% e 49%, respectivamente, quando utilizada em associação com Efavirenz. A importância clínica dessas alterações nos níveis plasmáticos da claritromicina não é conhecida. Ocorreu erupção cutânea em 46% dos voluntários não infectados que recebiam Efavirenz e claritromicina. Não é recomendado ajuste da dose de Efavirenz quando este for administrado com a claritromicina. Devem ser consideradas alternativas à claritromicina.
A coadministração de efavirenz (400 mg VO 1x/dia) com voriconazol (200 mg VO a cada 12 horas) em voluntários não-afetados em uma interação de dois modos. A AUC e a C máx de estado de equilíbrio do voriconazol diminuiu em 77% e 61%, respectivamente, enquanto AUC e a C máx de estado de equilíbrio do efavirenz aumentaram em 44% e 38%, respectivamente. A coadministração de doses padrões de efavirenz e voriconazol é contra-indicada.
Após a coadministração de efavirenz (300 mg VO 1x/dia) com voriconazol (300 mg 2x/dia) em voluntários nãoinfectados, a AUC e a C máx do voriconazol foram diminuídas em 55% e 36% respectivamente, em comparação com o voriconazol 200 mg 2x/dia isoladamente; a AUC do efavirenz foi equivalente, mas a C máx foi diminuída em 14% em comparação com efavirenz 600 mg 1x/dia isoladamente.
Após a coadministração de efavirenz (300 mg VO 1x/dia) com voriconazol (400 mg 2x/dia) em voluntários não infectados, a AUC do voriconazol foi diminuída em 7% e a C máx foi aumentada em 23% em comparação com o voriconazol 200 mg 2x/dia isoladamente. Estas diferenças não foram consideradas clinicamente significativas.
A AUC do efavirenz aumentou em 17% e a C máx foi equivalente em comparação com efavirenz 600 mg 1x/dia isoladamente.
Quando efavirenz é coadministrado com voriconazol, a dose de manutenção de voriconazol deve ser aumentada para 400 mg 2x/dia e a dose de efavirenz deve ser reduzida em 50%, i.e., para 300 mg 1x/dia. Quando o tratamento com voriconazol é interrompido, a posologia inicial de efavirenz deve ser restabelecida.
A coadministração de efavirenz (600 mg VO 1x/dia) com itraconazol (200 mg VO a cada 12 horas) em voluntários não-infectados diminuiu a AUC, C máx , e C mín de estado de equilíbrio do itraconazol em 39%, 37%, e 44%, respectivamente, e do hidroxiitraconazol em 37%, 35%, e 43%, respectivamente, em comparação com itraconazol administrado isoladamente. A farmacocinética do efavirenz não foi afetada. Como não se pode fazer nenhuma recomendação de dose para o itraconazol, deve ser considerado um tratamento antifúngico alternativo.
A coadministração de Efavirenz (400 mg VO 1x/dia) com posaconazol (400 mg VO 2x/dia) diminuiu a AUC e a C máx do posaconazol e, 50% e 45% respectivamente, em comparação com posaconazol administrado isoladamente. O uso concomitante de posaconazol e Efavirenz deve ser evitado a menos que o benefício supere o risco ao paciente.
A coadministração de efavirenz (600 mg, uma vez ao dia) com atovaquona e proguanil (250/100 mg, dose única) reduziu a AUC e a C máx em 75% e 44% para a atovaquona e a AUC em 43% para proguanil por meio da indução da glucoronidação. A administração concomitante de atovaquona/proguanil e efavirenz deve ser evitada sempre que possível.
A coadministração de efavirenz (600 mg, 1x/dia) com comprimidos de arteméter 20 mg/lumefantrina 120 mg (6 doses de 4 comprimidos por mais de três dias) resultou em diminuição da exposição (AUC) ao arteméter, di-hidroartemisinina (metabólito ativo do arteméter), e lumefantrina em aproximadamente 51%, 46% e 21%, respectivamente. A exposição ao efavirenz não foi significativamente afetada. Uma vez que concentrações reduzidas de arteméter, di-hidroartemisinina ou lumefantrina podem resultar em diminuição da eficácia de antimaláricos, recomenda-se precaução ao administrar Efavirenz concomitantemente com comprimidos de arteméter/lumefantrina.
A coadministração do efavirenz com inibidores da HMG-CoA reductase, atorvastatina , pravastatina, ou sinvastatina demonstrou reduzir a concentração plasmática da vastatina em voluntários não-infectados. Os níveis de colesterol devem ser periodicamente monitorados. Podem ser necessários ajustes de dose das vastatinas.
A coadministração de efavirenz (600 mg VO 1x/dia) com atorvastatina (10 mg VO 1x/dia) em voluntários não-infectados diminuiu a AUC e a C máx de estado de equilíbrio da atorvastatina em 43% e 12%, respectivamente, da 2-hidróxi atorvastatina em 35% e 13%, respectivamente, da 4-hidróxi atorvastatina em 4% e 47%, respectivamente, e dos inibidores ativos totais da HMG-CoA em 34% e 20%, respectivamente, em comparação com a atorvastatina administrada isoladamente.
A coadministração de efavirenz (600 mg VO 1x/dia) com pravastatina (40 mg VO 1x/dia) em voluntários não-infectados diminuiu a AUC e a C máx de estado de equilíbrio da pravastatina em 40% e 18%, respectivamente, em comparação com a pravastatina administrada isoladamente.
A coadministração de efavirenz (600 mg VO 1x/dia) com sinvastatina (40 mg VO 1x/dia) em voluntários não-infectados diminuiu a AUC e a C máx de estado de equilíbrio da sinvastatina em 69% e 76%, respectivamente, da sinvastatina ácida em 58% e 51%, respectivamente, dos inibidores totais ativos da HMGCoA redutase em 60% e 62%, respectivamente, e dos inibidores totais da HMG-CoA redutase em 60% e 70%, respectivamente, em comparação com a sinvastatina administrada isoladamente.
A coadministração de efavirenz com atorvastatina, pravastatina, ou sinvastatina não afetou os valores de AUC ou C máx do efavirenz. Não é necessário nenhum ajuste posológico para o efavirenz.
As concentrações plasmáticas e os efeitos potencialmente aumentaram ou diminuíram com Efavirenz.
A coadministração de efavirenz (600 mg VO 1x/dia) com carbamazepina (400 mg 1x/dia) em voluntários não-infectados resultou em uma interação de dois modos. A AUC, C máx e C mín de estado de equilíbrio da carbamazepina diminuiu em 27%, 20% e 35%, respectivamente, ao passo que a AUC, C máx e C mín de estado de equilíbrio do efavirenz foram reduzidas em 36%, 21%, e 47%, respectivamente. A AUC, C máx e C mín de estado de equilíbrio do metabólito ativo carbamazepina epóxido permaneceram inalterados. Os níveis plasmáticos da carbamazepina devem ser monitorados periodicamente. Não há nenhum dado sobre a coadministração de doses mais altas de qualquer um dos medicamentos; portanto, nenhuma recomendação posológica pode ser feita, e deve ser considerado um tratamento anticonvulsivante alternativo.
Não há nenhum dado disponível sobre potenciais interações do efavirenz com fenitoína , fenobarbital , ou outros anticonvulsivantes que são substratos das isoenzimas CYP450. Quando o efavirenz é administrado concomitantemente com estes agentes, existe a possibilidade de redução ou aumento das concentrações plasmáticas de cada medicamento; portanto, deve ser realizado monitoramento periódico dos níveis plasmáticos.
Estudos específicos de interação não foram realizados com efavirenz e vigabatrina ou gabapentina . Não são esperadas interações clinicamente significativas uma vez que a vigabatrina e a gabapentina são eliminadas exclusivamente inalteradas na urina e seria improvável que competisse pelas mesmas enzimas metabólicas e vias de eliminação do efavirenz.
Quando um contraceptivo oral (etinil estradiol 0,035 mg/norgestimato 0,25 mg 1x/dia) e efavirenz (600 mg 1x/dia) foi coadministrado por 14 dias, o efavirenz não apresentou nenhum efeito sobre as concentrações de etinil estradiol, porém as concentrações plasmáticas de norelgestromina e levonorgestrel , metabólitos ativos do norgestimato, foram acentuadamente diminuídas na presença do efavirenz (64%, 46%, e 82% de redução da AUC, C máx e C mín , respectivamente da norelgestromina, e redução de 83%, 80%, e 86% da AUC, C máx , e C mín , respectivamente do levonorgestrel). A significância clínica destes efeitos não é conhecida.
Não se observou nenhum efeito de etinil estradiol / norgestimato sobre as concentrações plasmáticas do efavirenz.
Existem informações limitadas a respeito do efavirenz e contraceptivos hormonais injetáveis. Em um estudo de interação medicamentosa de três meses do acetato de depo-medroxiprogesterona (DMPA) e efavirenz, os níveis plasmáticos de progesterona para todos os indivíduos permaneceram abaixo de 5 ng/ml, consistente com a supressão da ovulação.
A interação entre etonogestrel e efavirenz não foi estudada. Pode-se esperar uma exposição diminuída ao etonogestrel (indução da CYP3A4), e houve relatos ocasionais de farmacovigilância de falha contraceptiva com etonogestrel em pacientes expostos ao efavirenz.
Quando é administrado um imunossupressor metabolizado pela CYP3A4 (por exemplo: ciclosporina , tacrolimo , ou sirolimo ) com efavirenz, uma redução da exposição ao imunossupressor pode ocorrer devido à indução da CYP3A4. Podem ser requeridos ajustes de dose do imunossupressor. Ao iniciar ou suspender o tratamento com efavirenz, é recomendado acompanhamento da concentração de imunossupressor por pelo menos 2 semanas (até que a concentração estável seja atingida).
Em um estudo com usuários de drogas injetáveis por via endovenosa infectados pelo HIV, a administração concomitante de efavirenz e metadona resultou em redução dos níveis plasmáticos da metadona e em sinais de abstinência de opiáceos. A dose de metadona foi aumentada em 22%, em média, para aliviar esses sintomas. Sinais de abstinência devem ser monitorados nesses pacientes e a dose de metadona deve ser aumentada conforme necessário para aliviar esses sintomas.
Não houve efeitos clinicamente significativos nos parâmetros farmacocinéticos quando a paroxetina e o efavirenz foram administrados concomitantemente. Não é necessário ajuste de dose tanto para o efavirenz como para a paroxetina quando esses medicamentos são administrados concomitantemente. A sertralina não alterou significativamente a farmacocinética do efavirenz. O efavirenz reduziu a C máx , a C24 e a AUC da sertralina em 28,6%-46,3%. A dose de sertralina deve ser aumentada quando administrada com o efavirenz para compensar a redução do metabolismo da sertralina pelo efavirenz. O aumento da dose da sertralina deve ser monitorado de acordo com a resposta clínica. A bupropiona (150 mg, dose única, liberação sustentada) quando administrada com efavirenz (600 mg, uma vez ao dia) reduziu a AUC e C máx em 55% e 34% respectivamente. A AUC da hidroxibupropiona não foi alterada e a C máx aumentou em 50% por meio da indução da CYP2B6.
Aumentos na dose de bupropiona devem ser orientados pela resposta clínica, mas não devem exceder a dose máxima recomendada. Não é necessário ajuste da dose de efavirenz.
A cetirizina não exerceu efeito clinicamente significativo nos parâmetros farmacocinéticos do efavirenz. O efavirenz reduziu a C máx da cetirizina para 24%, mas não alterou a AUC desse medicamento. Não se espera que essas alterações sejam clinicamente significativas. Ajustes de dose não são necessários nem para o efavirenz nem para a cetirizina quando esses medicamentos são administrados concomitantemente.
O efavirenz aumentou a C máx e a AUC do lorazepam para 16,3% e 7,3%, respectivamente. É improvável que a interação farmacocinética do efavirenz com o lorazepam seja clinicamente significativa. Não são necessários ajustes de dose nem para o efavirenz nem para o lorazepam quando esses medicamentos são administrados concomitantemente.
Bloqueadores do canal de cálcio: a coadministração de efavirenz (600 mg VO 1x/dia) com diltiazem (240 mg VO 1x/dia) em voluntários não-infectados reduziu a AUC, C máx , e C mín de estado de equilíbrio do diltiazem em 69%, 60%, e 63%, respectivamente; desacetil diltiazem em 75%, 64%, e 62%, respectivamente; e N-monodesmetil diltiazem em 37%, 28%, e 37%, respectivamente, em comparação com diltiazem administrado isoladamente. Os ajustes de dose de diltiazem devem ser orientados pela resposta clínica (consultar a bula do produto para diltiazem).
Embora os parâmetros farmacocinéticos de efavirenz tenham sido discretamente aumentados (11% -16%), estas alterações não são consideradas clinicamente significativas e, portanto, não é necessário nenhum ajuste de dose para efavirenz quando administrado com diltiazem.
Não está disponível nenhum dado sobre as potenciais interações de efavirenz com outros bloqueadores do canal de cálcio que são substratos da enzima CYP3A4 (p.ex., verapamil , felodipina, nifedipina , nicardipina). Quando efavirenz é administrado concomitantemente com um destes agentes, existe um potencial para redução das concentrações plasmáticas do bloqueador de canal de cálcio. Os ajustes de dose devem ser orientados pela resposta clínica (consulte a bula do fabricante correspondente para o bloqueador de canal de cálcio).
O efavirenz não se liga aos receptores de canabinóide. Foram relatados em exames de triagem resultados falso-positivos de exames de urina para canabinóide em voluntários não infectados e infectados pelo HIV que receberam Efavirenz.É recomendada a confirmação de resultados de exames de triagem positivos para canabinóides por um método mais específico, como a cromatografia gasosa/espectrometria de massa.
Precauções
Efavirenz não deve ser usado como agente único para tratar a infecção causada pelo HIV ou adicionado como agente único a um esquema que tenha falhado.
Ao prescrever medicamentos que serão utilizados concomitantemente com Efavirenz, os médicos devem consultar as respectivas bulas emitidas pelos fabricantes.
Quando qualquer medicação antirretroviral em esquema combinado for interrompida por suspeita de intolerância, deve-se considerar seriamente a descontinuação simultânea de todas as medicações antirretrovirais. As medicações antirretrovirais devem ser reiniciadas ao mesmo tempo, quando desaparecerem os sintomas de intolerância. A monoterapia intermitente e a reintrodução sequencial de agentes antirretrovirais não são aconselháveis por causa do aumento da possibilidade de seleção de vírus mutantes resistentes aos medicamentos.
A coadministração de Efavirenz com associações de medicamentos que contêm efavirenz não é recomendada, a menos que seja necessária para ajuste de dose (exemplo: com rifampina).
Foram observadas malformações em fetos de animais tratados com o efavirenz; portanto, a gravidez deve ser evitada por mulheres que estejam recebendo Efavirenz. Métodos anticoncepcionais por barreiras devem ser sempre adotados em combinação com outros métodos anticoncepcionais (por exemplo, anticoncepcionais orais ou outros anticoncepcionais hormonais).
As concentrações plasmáticas de efavirenz podem ser alteradas por substratos, inibidores ou indutores da CYP3A4. Do mesmo modo, o efavirenz pode alterar as concentrações plasmáticas de medicamentos metabolizados pela CYP3A4 ou CYP2B6. O efeito proeminente de efavirenz no estado de equilíbrio é a indução da CYP3A4 e CYP2B6. No entanto, o efavirenz demonstrou ter efeitos inibitórios da CYP3A4 in vitro ; portanto, existe o potencial teórico de que os níveis dos medicamentos sejam aumentados temporariamente para agentes metabolizados pela CYP3A4. Deve-se ter precaução durante os primeiros dias de tratamento com Efavirenz para pacientes que estejam tomando substratos da CYP3A4 que tenham índice terapêutico estreito e um potencial para reações adversas graves e/ou que ameacem a vida (por exemplo: arritmias cardíacas, sedação prolongada ou depressão respiratória). Devem ser tomadas precauções para agentes tais como derivados da ergô (di-hidroergotamina, ergonovina, ergotamina, metilergonovina), midazolam, triazolam, bepridil, cisaprida e pimozida .
Foi relatada erupção cutânea leve a moderada em estudos clínicos com Efavirenz, a qual geralmente desaparece com a continuação do tratamento. Anti-histamínicos e/ou corticosteroides adequados podem melhorar a tolerabilidade e acelerar o desaparecimento da erupção cutânea. Foi relatada erupção cutânea grave, com vesiculação, descamação úmida ou ulceração, em menos de 1% dos pacientes tratados com Efavirenz. A incidência de eritema polimorfo ou de síndrome de Stevens-Johnson foi de 0,14%. Efavirenz deve ser descontinuado pelos pacientes que desenvolverem erupção cutânea grave com vesiculação, descamação, acometimento de mucosas ou febre . Efavirenz não é recomendado para pacientes que tenham apresentado reações cutâneas com risco de vida (como por exemplo Síndrome de Stevens-Johnson). Se o tratamento com Efavirenz for descontinuado, deve-se também considerar a interrupção do tratamento com outros agentes antirretrovirais, a fim de evitar o desenvolvimento de vírus resistentes ao medicamento.
Foi relatada erupção cutânea em 58 de 182 crianças (32%) tratadas com Efavirenz em três estudos clínicos por uma mediana de 123 semanas. A erupção cutânea foi considerada de intensidade grave em seis pacientes. A mediana do tempo para início da erupção cutânea em pacientes pediátricos foi de 27 dias (intervalo de 3-1504 dias). Antes do início do tratamento com Efavirenz em crianças, pode-se considerar a profilaxia com antihistamínicos adequados.
Foram relatadas experiências adversas psiquiátricas em pacientes tratados com efavirenz. Os pacientes com histórico de distúrbios psiquiátricos parecem apresentar maior risco de experiências adversas psiquiátricas graves. Há relatos observados por farmacovigilância, de morte por suicídio, alucinações e comportamentos psicóticos; no entanto, a relação causal com o uso do efavirenz não pode ser determinada. Caso apresentem esses sintomas, os pacientes devem ser alertados a procurar imediatamente seu médico para avaliar a possibilidade de esses sintomas estarem relacionados com o uso do efavirenz e, se estiverem, para determinar se o risco de continuar o tratamento supera os benefícios.
Em estudos clínicos com pacientes recebendo diariamente 600 mg de efavirenz, foram relatados frequentemente como efeitos indesejáveis os seguintes sintomas: tontura, insônia, sonolência, dificuldade de concentração e padrão anormal de sonhos. Os sintomas neurológicos geralmente iniciam-se durante o primeiro ou o segundo dia de tratamento e geralmente melhoram depois das primeiras 2 a 4 semanas. Os pacientes devem ser informados de que esses sintomas comuns provavelmente melhorarão no decorrer do tratamento e não indicam início subsequente de qualquer um dos sintomas psiquiátricos menos frequentes.
Foram observadas convulsões raramente em pacientes recebendo efavirenz, geralmente na presença de histórico médico conhecido de convulsões. Pacientes que estejam recebendo medicamentos anticonvulsivantes concomitantes metabolizados principalmente pelo fígado , como a fenitoína, carbamazepina e fenobarbital podem necessitar de monitoramento periódico dos níveis plasmáticos. Em um estudo de interação medicamentosa, as concentrações plasmáticas de carbamazepina foram diminuídas quando a carbamazepina foi coadministrada com efavirenz. Deve-se ter cuidado com qualquer paciente que apresente histórico de convulsões.
A síndrome de reconstituição imunológica tem sido relatada em pacientes tratados com a terapia de combinação antirretroviral (TCAR), incluindo Efavirenz. Durante a fase inicial do tratamento, um paciente cujo sistema imunológico responda à TCAR pode desenvolver uma resposta inflamatória a infecções oportunistas insensíveis ou residuais, as quais podem necessitar de avaliação e tratamento adicional.
Foram também reportados distúrbios autoimunes (como Doenças de Graves) durante o início da reconstituição imune, no entanto, o tempo reportado para início é variável e estes eventos podem ocorrer muitos meses após o início do tratamento.
Tendo em vista o metabolismo extensivo do efavirenz mediado pelo citocromo P450 e a limitada experiência clínica em pacientes com doença hepática crônica, deve-se ter cautela ao administrar Efavirenz a pacientes com doença hepática.
Pacientes com hepatite crônica B ou C e tratados com a terapia de combinação antirretroviral estão em risco aumentado de eventos adversos hepáticos graves e potencialmente fatais. Alguns dos relatos de farmacovigilância de insuficiência hepática ocorreram em pacientes sem nenhuma doença hepática preexistente ou outros fatores de risco identificáveis. O monitoramento de enzimas hepáticas também deve ser considerado para pacientes sem disfunção hepática preexistente ou outros fatores de risco.
A farmacocinética do efavirenz não foi estudada em pacientes com insuficiência renal, entretanto menos de 1% de uma dose de efavirenz é excretada inalterada na urina; portanto, o impacto do comprometimento renal na eliminação do efavirenz deve ser mínimo.
Não há experiência de uso de Efavirenz em pacientes com insuficiência renal grave e é recomendado monitoramento de segurança nesta população.
Para pacientes com histórico de hepatite B ou C ou nos quais se suspeita de presença dessas infecções e para pacientes tratados com outras medicações associadas à toxicidade hepática, recomenda-se a monitoração das enzimas hepáticas. No caso de pacientes com elevações persistentes das transaminases séricas cinco vezes acima do limite superior da normalidade, o benefício do tratamento contínuo com Efavirenz deve ser contraposto aos riscos desconhecidos de toxicidade hepática significativa.
Gravidez: Categoria D.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
A gravidez deve ser evitada por mulheres que estejam sendo tratadas com efavirenz. Métodos anticoncepcionais por barreiras devem ser sempre adotados em combinação com outros métodos anticoncepcionais (por exemplo: anticoncepcionais orais ou outros anticoncepcionais hormonais).
Devido à meia-vida longa do efavirenz, o uso de medidas contraceptivas adequadas por 12semanas após a descontinuação de Efavirenz é recomendado. Mulheres com potencial para engravidar devem realizar um teste de gravidez antes do início da terapia com efavirenz. O efavirenz não deve ser utilizado durante a gravidez a menos que o benefício potencial para a mãe supere claramente o risco potencial ao feto e não existam outras opções de tratamento apropriadas. Se uma mulher tomar efavirenz durante o primeiro trimestre de gravidez ou engravidar enquanto estiver tomando efavirenz, ela deve ser informada do potencial prejuízo ao feto.
Não existem estudos adequados e bem-controlados do efavirenz em mulheres grávidas. Na experiência de farmacovigilância por meio de um registro de gravidez antirretroviral, mais de 700 casos de gravidez com exposição no primeiro trimestre ao efavirenz como parte de uma terapia de combinação antirretroviral foram relatados sem nenhum padrão específico de malformação. Neste registro, um pequeno número de casos de defeitos do tubo neural, incluindo meningomielocele, foi relatado; a maioria destes relatos foi retrospectiva e a causalidade não foi estabelecida.
Efavirenz é secretado no leite de ratas lactantes e foi demonstrado que o efavirenz passa através do leite humano. É recomendado às mães que estejam tomando efavirenz não amamentarem seus filhos. Recomenda-se que mulheres HIV-positivas não amamentem sob quaisquer circunstâncias, a fim de evitar a transmissão do HIV.
Foi avaliado um número insuficiente de pacientes idosos em estudos clínicos para determinar se eles reagem de modo diferente em relação aos pacientes mais jovens.
Efavirenz não foi avaliado em crianças com menos de 3 anos de idade ou com peso inferior a 13 kg. Existem evidências indicando que a farmacocinética do efavirenz pode ser alterada em crianças muito novas; por essa razão, não se deve administrar a solução oral de efavirenz a crianças com menos de 3 anos de idade. Os comprimidos de Efavirenz não são adequados para crianças com peso inferior a 40 kg; para esses pacientes, está disponível a solução oral de Efavirenz.
Efavirenz pode causar tontura, comprometimento da concentração e/ou sonolência.
Os pacientes devem ser orientados de que se apresentarem estes sintomas, eles deverão evitar tarefas potencialmente perigosas, como dirigir ou operar máquinas.
Atenção: o uso incorreto causa resistência do vírus da aids e falha no tratamento.
Resultados de Eficácia
Nos estudos clínicos descritos a seguir, a principal medida de eficácia foi a porcentagem de pacientes com níveis plasmáticos de RNA do HIV <400 cópias/mL, medidos pelo ensaio monitor de HIV-1 (Amplicor®) PCR-TR. O limite inferior de quantificação desse ensaio é mais baixo do que 400 cópias/mL; portanto, ficou estabelecido que, para a análise da alteração média em relação ao período basal, valores abaixo do limite de quantificação seriam considerados 400 cópias/mL. A quantificação de RNA do HIV também foi obtida por um ensaio de PCRTR, cujo limite inferior era de 50 cópias/mL (ultrassensível).
Nas análises NC=F (pacientes que não completaram o tratamento e os quais foram considerados falha terapêutica) apresentadas, foram consideradas falhas terapêuticas quando os pacientes terminaram o estudo precocemente por qualquer razão ou para os quais faltava um resultado de RNA do HIV, precedido ou seguido de um resultado acima do limite de quantificação do ensaio (>400 cópias/mL). Nas análises de dados observados, apresentadas nas tabelas abaixo, considerou-se falha terapêutica quando, no ponto de tempo especificado, os pacientes sob tratamento apresentavam RNA do HIV >400 cópias/mL.
O estudo 006 foi um estudo randômico e aberto para avaliar a supressão de RNA do HIV no plasma por Efavirenz em combinação com o indinavir (IDV) ou com a associação zidovudina (AZT) + lamivudina (3TC) em comparação com a combinação indinavir + zidovudina + lamivudina em pacientes infectados pelo HIV não tratados anteriormente com lamivudina, inibidores da protease e ITRNNs. Os pacientes receberam um dos três esquemas de tratamento: Efavirenz (600 mg uma vez ao dia) + indinavir (1.000 mg a cada 8 horas) ou Efavirenz (600 mg uma vez ao dia) + zidovudina (300 mg a cada 12 horas) + lamivudina (150 mg a cada 12 horas) versus indinavir (800 mg a cada 8 horas) + zidovudina (300 mg a cada 12 horas) + lamivudina (150 mg a cada 12 horas). São apresentadas as análises referentes aos dados de 48 semanas de 614 pacientes (média de idade de 36,3 anos [variação de 18 a 64 anos], 58% caucasianos e 86% homens). No período basal, o número médio de células CD4 era de 342 células/mm 3 e o nível plasmático médio de RNA do HIV, 4,76 log 10 cópias/mL. A figura 1 apresenta a análise NC=F da porcentagem de pacientes que atingiram níveis plasmáticos de RNA do HIV <400 cópias/mL. A tabela 1 apresenta, de forma resumida, outros resultados de eficácia.
Figura 1
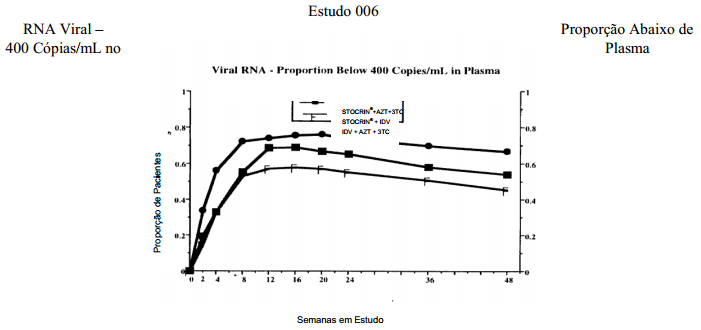
Tabela 1. Estudo 006 – Resumo dos Principais Resultados de Eficácia – 48 a Semana
Efavirenz + AZT + 3TC
Total de pacientes (distribuição randômica)
20
Observados (n/N)
96,9% (126/130) *
(IC 95%)
(94,0, 99,9)
Observados (n/N)
90,7% (117/129) *
(IC 95%)
(85,7, 95,7
NC=F (n/N)
61,6% (117/190) *
(IC 95%)
(54,7, 68,5)
Amplicor
-1,98 (0,05) *, **, ***
Ultrassensível
-2,32 (0,09) *, **, ***
187,04 (11,8) *, ***
* Diferença estatisticamente significativa (p< 0,05) entre Efavirenz +AZT+3TC e IDV+AZT+3TC.
** Diferença estatisticamente significativa (p< 0,05) entre os grupos de tratamento.
*** Diferença estatisticamente significativa (p< 0,05) em relação ao período basal.
† Diferença estatisticamente significativa (p< 0,05) entre Efavirenz +IDV e IDV+AZT+3TC.
IC: Intervalo de Confiança.
NC=F: Não Completou o Estudo = Falha Terapêutica.
O estudo 020 foi um estudo randômico, duplo-cego e controlado com placebo, que envolveu pacientes já tratados com ITRN e não tratados anteriormente com inibidor de protease e ITRNNs, para comparar o tratamento quádruplo com Efavirenz + indinavir + 2 inibidores da transcriptase reversa análogos de nucleosídeos versus tratamento triplo com indinavir + 2 ITRNs após 24 semanas de tratamento. Os pacientes receberam Efavirenz (600 mg uma vez ao dia) + indinavir (1.000 mg a cada 8 horas) + 2 ITRNs ou indinavir (800 mg a cada 8 horas) + 2 ITRNs. Dos 327 pacientes (média de idade de 38,5 anos [variação de 20 a 69 anos], 52% caucasianos, 83% homens), 69% mudaram seus esquemas de ITRN no início do estudo. No período basal, o número médio de células CD4 era de 328 células/mm 3 e o nível médio de RNA do HIV no plasma, 4,41 log 10 cópias/mL. A figura 2 mostra a análise NC=F da porcentagem de pacientes que atingiram níveis plasmáticos de RNA do HIV <400 cópias/mL na 24a semana. A tabela 2 apresenta, de forma resumida, outros resultados de eficácia.
Figura 2

Tabela 2. Estudo 020 – Resumo dos Principais Resultados de Eficácia – 24 a Semana
Efavirenz + IDV + AZT + 3TC
Total de pacientes (distribuição randômica)
157
Observados (n/N)
83,0% (93/112) *
(IC 95%)
(75,6; 90,4)
Ensaio ultrassensível
Observados (n/N)
68,8% (77/112)
(IC 95%)
(59,7; 77,8)
NC=F (n/N)
49,4% (77/156) *
(IC 95%)
(41,2; 57,5)
Amplicor
-1,45 (0,08) *, **
Ultrassensível
-2,25 (0,10) *, **
104 (9,1) *, **
* Diferença estatisticamente significativa (p< 0,05) entre os grupos de tratamento.
** Alteração média estatisticamente significativa (p< 0,05) em relação ao período basal.
IC: Intervalo de Confiança.
NC=F: Não Completou o Estudo = Falha Terapêutica.
O estudo ACTG 364 foi um estudo com duração de 48 semanas, duplo-cego, randômico, controlado com placebo, que envolveu 196 pacientes infectados pelo HIV já tratados com ITRN (média de idade de 41 anos [variação de 18 a 76 anos], 74% caucasianos, 88% homens). Os pacientes receberam ITRNs em combinação com Efavirenz (600 mg uma vez ao dia) ou nelfinavir (NFV; 750 mg três vezes ao dia), ou Efavirenz (600 mg uma vez ao dia) + nelfinavir. Ao serem admitidos no estudo, todos os pacientes foram designados para um novo esquema aberto com ITRN, dependendo da sua experiência anterior de tratamento com esses antirretrovirais. A tabela 3 apresenta, de forma resumida, os resultados globais de eficácia.
Tabela 3. ACTG 364 – Resumo dos Principais Resultados de Eficácia – 48 a Semana
Efavirenz + NFV + ITRNs
Total de pacientes (distribuição randômica)
65
Observados (n/N)
82,1% (46/56) *, ††
(IC 95%)
(72,1; 92,2)
NC=F
70,3% (45/64) *, ††
(IC 95%)
(59,1; 81,5)
Amplicor (Observados)
-0,96 (0,11) **
107 (17,9) **
* Diferença estatisticamente significativa (p< 0,05) entre os grupos de tratamento.
** Diferença estatisticamente significativa (p< 0,05) em relação ao período basal.
*** Diferença estatisticamente significativa entre os grupos com NFV e com Efavirenz (p < 0,05).
† As análises excluem os dados de acompanhamento obtidos após o término do tratamento randômico.
†† Diferença estatisticamente significativa entre os grupos com NFV e com Efavirenz + NFV (p < 0,05).
IC: Intervalo de Confiança.
NC=F: Não Completou o Estudo = Falha Terapêutica.
Em uma fase-piloto desse estudo, 32 pacientes (75% já tratados com ITRN) foram designados de modo randômico para receber placebo (10) ou monoterapia (22) com 200 mg de Efavirenz uma vez ao dia durante duas semanas. Em dois grupos que receberam 200 mg de Efavirenz uma vez ao dia como monoterapia durante duas semanas, os níveis plasmáticos médios de RNA do HIV medidos pelo ensaio Amplicor foram reduzidos para 1,67 log 10 cópias/mL em relação a 5,02 log 10 cópias/mL no período basal (98% de supressão) e para 1,52 log 10 cópias/mL em relação a 5,21 log 10 cópias/mL no período basal (97% de supressão), respectivamente. Observou-se aumento de células CD4 de 98 ± 57,5 células/mm 3 ; no grupo placebo não houve alteração. Em uma segunda fase deste estudo, 59 pacientes (63% já tratados com ITRN, 19% já tratados com lamivudina) foram designados de modo randômico para receber Efavirenz (200 mg uma vez ao dia, aumentados a seguir para 600 mg uma vez ao dia) e indinavir (800 mg ou 1.000 mg a cada 8 horas, aumentados a seguir para 1.000 mg a cada 8 horas para todos os pacientes). Entre os pacientes designados de modo randômico para receber Efavirenz mais indinavir, a proporção de pacientes com RNA do HIV <400 cópias/mL na análise NC=F foi de 67,2% na 72 a semana. A taxa de resposta foi semelhante, independentemente de tratamento anterior com ITRN.
Estudo duplo-cego, controlado com placebo, de variação de dose, que avaliou a segurança e a eficácia de Efavirenz em combinação com a zidovudina e a lamivudina em 137 pacientes infectados pelo HIV-1 não tratados anteriormente com antirretroviral. No período basal, o número médio de células CD4 era de 367 células/mm 3 e o RNA do HIV no plasma, 4,72 log 10 cópias/mL. Os pacientes foram designados de modo randômico para receber 200 mg, 400 mg ou 600 mg de Efavirenz ou placebo equivalente em combinação com zidovudina (300 mg duas vezes ao dia) e lamivudina (150 mg duas vezes ao dia). Na 16a semana, a taxa de resposta observada (porcentagem <400 cópias de RNA do HIV/mL) nos grupos tratados com Efavirenz foi significativamente mais alta do que nos grupos do placebo (zidovudina + lamivudina) e variou de 88,9% a 93,5% em todos os grupos de tratamento com Efavirenz versus 44,4% no grupo controle. A análise NC=F da taxa de resposta foi significativamente superior à do placebo e variou de 72,7% a 80,6% em todos os grupos de tratamento com Efavirenz versus 36,4% no grupo controle. O número de células CD4 aumentou significativamente em todos os grupos de tratamento; não foram observadas diferenças estatisticamente significativas entre os quatro grupos de tratamento.
Em uma extensão de longo prazo do estudo 005, em andamento, todos os 11 pacientes originalmente designados de modo randômico para receber 600 mg de Efavirenz – cujos níveis plasmáticos de RNA do HIV situavam-se abaixo do limite de detecção na 16a semana e para os quais estão disponíveis resultados de 36 semanas – mantiveram níveis plasmáticos de RNA do HIV inferiores a 400 cópias/mL.
O estudo ACTG 382 é um estudo em andamento, aberto, com 48 semanas de duração, envolvendo 57 pacientes pediátricos já tratados com ITRN, para caracterizar a farmacocinética, a atividade antirretroviral e a segurança de Efavirenz em combinação com nelfinavir (20-30 mg/kg três vezes ao dia) e ITRNs. A média de idade é de oito anos (variação de 3 a 16 anos). Os níveis plasmáticos médios de RNA do HIV no período basal eram de 4,09 log 10 ( + 0,69) cópias/mL. A dose inicial de Efavirenz foi de 600 mg uma vez ao dia, ajustada pela superfície corpórea, com o objetivo de obter níveis de AUC entre 190-380 µM•h. Nos pacientes que completaram 48 semanas de tratamento, a taxa de respondentes com RNA do HIV <400 cópias/mL foi de 60% (34/57). O número médio de células CD4 aumentou em 63 células/mm 3 em relação ao período basal.
Características Farmacológicas
O efavirenz é um inibidor seletivo não nucleosídeo da transcriptase reversa do vírus da imunodeficiência humana tipo 1 (HIV-1). O efavirenz é um inibidor não competitivo da transcriptase reversa (TR) do HIV-1 no que diz respeito à matriz e trifosfatos básicos ou nucleosídeos, com um pequeno componente de inibição competitiva. A transcriptase reversa do HIV tipo 2 e as DNA polimerases α, β, γ e δ de células humanas não são inibidas por concentrações do efavirenz muito acima daquelas atingidas clinicamente.
Concentrações plasmáticas máximas de 1,6-9,1 µM de efavirenz foram alcançadas cerca de 5 horas após doses únicas de 100 mg a 1.600 mg, administradas por via oral a voluntários não infectados. Com doses de até 1.600 mg, foram observados aumentos da C máx e da AUC relacionados à dose; os aumentos não chegaram a ser proporcionais, sugerindo redução da absorção com doses mais altas. O tempo até a obtenção de concentrações plasmáticas máximas (3-5 horas) não foi alterado após administração múltipla e as concentrações plasmáticas em estado de equilíbrio foram alcançadas em 6-7 dias.
Em pacientes infectados pelo HIV-1, a C máx , a C mín e a AUC médias em estado de equilíbrio foram lineares com doses de 200 mg, 400 mg e 600 mg ao dia. Em 35 pacientes que receberam 600 mg de Efavirenz uma vez ao dia, a C máx e a C mín em estado de equilíbrio foram de 12,9 µM e de 5,6 µM, respectivamente, e a AUC foi de 184 µM•h.
Em voluntários adultos não infectados, a C máx e a AUC de uma dose de 240 mg da solução oral de Efavirenz foram 78% e 97%, respectivamente, dos valores obtidos quando Efavirenz foi administrado em cápsulas duras de 200 mg.
Efeito de alimentos na absorção oral: a biodisponibilidade de uma dose única de 600 mg de efavirenz em voluntários não infectados aumentou 22% e 17%, respectivamente, quando administrada com uma refeição com alto teor de gordura ou com uma refeição de composição normal em relação à biodisponibilidade da dose de 600 mg administrada em jejum. Efavirenz pode ser administrado com ou sem alimentos.
O efavirenz liga-se intensamente (aproximadamente 99,5%-99,75%) às proteínas plasmáticas humanas, principalmente à albumina. Em pacientes infectados pelo HIV-1 (N= 9) que receberam 200 mg a 600 mg de Efavirenz uma vez ao dia durante pelo menos um mês, as concentrações no líquor variaram de 0,26% a 1,19% (média de 0,69%) da concentração plasmática correspondente. Essa proporção é aproximadamente três vezes maior do que a fração não ligada às proteínas (livre) de efavirenz no plasma.
Estudos em seres humanos e estudos in vitro nos quais foram utilizados microssomos hepáticos humanos demonstraram que o efavirenz é metabolizado principalmente pelo sistema do citocromo P450 em metabólitos hidroxilados, que sofrem glicuronidação subsequente. Esses metabólitos são essencialmente inativos contra o HIV-1. Os estudos in vitro sugerem que a CYP3A4 e a CYP2B6 são as principais isoenzimas responsáveis pelo metabolismo do efavirenz. Os estudos in vitro mostraram que o efavirenz inibiu as isoenzimas CYP 2C9, 2C19 e 3A4 com valores de Ki (8,5-17 µM) na faixa das concentrações de efavirenz observadas no plasma. Em estudos in vitro , o efavirenz não inibiu a CYP2E1 e inibiu a CYP2D6 e a CYP1A2 (valores de Ki de 82-160 µM) apenas em concentrações muito acima das alcançadas clinicamente.
A exposição plasmática de efavirenz pode ser aumentada em pacientes com a variante genética homozigota G516T da isoenzima CYP2B6. As implicações clínicas dessa associação são desconhecidas; no entanto, o potencial para um aumento de freqüência e gravidade de eventos adversos associados ao efavirenz não pode ser excluído.
O efavirenz demonstrou induzir as enzimas do citocromo P450, o que resulta na indução do seu próprio metabolismo. Doses múltiplas de 200-400 mg ao dia durante 10 dias resultaram em acúmulo menor do que o previsto (22%-42% mais baixo) e em meia-vida terminal mais curta, de 40-55 horas (meia-vida da dose única: de 52-76 horas). Espera-se que o grau de indução da CYP3A4 seja semelhante entre uma dose de 400 mg e outra de 600 mg de efavirenz com base nos estudos de interação farmacocinética, nos quais doses diárias de 400 mg ou 600 mg de efavirenz, em combinação com o indinavir, não pareceram causar redução adicional da AUC do indinavir em comparação com uma dose de 200 mg de efavirenz.
O efavirenz tem meia-vida terminal relativamente longa: de 52 a 76 horas após doses únicas e de 40 a 55 horas após doses múltiplas. Aproximadamente 14%-34% de uma dose de efavirenz marcado radioativamente foi recuperada na urina e menos de 1% da dose foi excretada na urina como efavirenz inalterado.
O significado clínico da sensibilidade do HIV-1 ao efavirenz in vitro não foi estabelecido. A atividade antiviral do efavirenz in vitro foi avaliada em linhagens celulares linfoblastóides, em células mononucleares do sangue periférico (CMSPs) e em culturas de macrófagos/monócitos enriquecidas a partir de CMSPs. A concentração inibitória de 90%-95% (CI90-95) do efavirenz para cepas do tipo selvagem adaptadas em laboratório e para isolados clínicos variou de 1,7 a ≤25 nM. A potência do efavirenz contra variantes com mutações de S48T, V108I, V179D, Y181C, P236L ou variantes com substituições de aminoácidos no gene da protease foi semelhante à observada contra o tipo selvagem. Foi observada resistência modesta (inferior a nove vezes) contra variantes com as mutações A98G, K101E, V106A, Y188C ou G190A. As mutações pontuais que levaram à resistência mais alta aparente à inibição pelo efavirenz in vidro foram L100I (resistência de 17 a 22 vezes) e K103N (resistência de 18 a 33 vezes). As seguintes variantes de múltiplos pares de base, que sofreram mutação que codificam as TRs com uma ou mais substituições de aminoácidos, mostraram aumento da resistência ao efavirenz in vitro em relação ao tipo selvagem: S48T+G190S (97 vezes), Y181C+K103N (133 vezes), G190A+K103N (130 vezes), Y188L (140 a 500 vezes), K101E+K103N (500 vezes) e L100I+K103N (>1.000 vezes).
O efavirenz demonstrou atividade sinérgica em cultura celular em associação com os inibidores da transcriptase reversa análogos de nucleosídeos (ITRNs), zidovudina (AZT) ou didanosina (ddl), ou com o indinavir, um inibidor da protease.
A potência do efavirenz em cultura celular contra variantes virais com substituições de aminoácidos nas posições 48, 108, 179, 181 ou 236 na TR ou variantes com substituições de aminoácidos na protease foi semelhante à observada contra cepas virais do tipo selvagem. As substituições isoladas que levaram à resistência mais alta ao efavirenz em cultura celular correspondem a uma alteração leucina-para-isoleucina na posição 100 (L100I, resistência de 17 a 22 vezes) e uma lisina-para-asparagina na posição 103 (K103N, resistência de 18 a 33 vezes). Foi observada perda de sensibilidade superior a 100 vezes contra variantes do HIV que expressam K103N além de outras substituições de aminoácidos na TR. K103N foi a substituição na TR mais frequentemente observada em isolados virais de pacientes que apresentaram rebote significativo da carga viral durante estudos clínicos que avaliaram o efavirenz em combinação com o indinavir ou a associação zidovudina + lamivudina. Também foram observadas substituições nas posições 98, 100, 101, 108, 138, 188, 190 ou 225 da TR, porém em menor frequência e muitas vezes somente em combinação com K103N. O padrão de substituição de aminoácidos na TR associado com resistência ao efavirenz foi independente de outras medicações antivirais usadas em combinação com o efavirenz.
Os perfis de resistência cruzada para o efavirenz, a nevirapina e a delavirdina em cultura celular mostraram que a substituição de K103N confere perda de sensibilidade aos três inibidores da transcriptase reversa não nucleosídeos (ITRNNs). Dois de três isolados clínicos resistentes à delavirdina examinados apresentavam resistência cruzada ao efavirenz e continham a substituição de K103N. Um terceiro isolado com substituição na posição 236 da TR não apresentou resistência cruzada ao efavirenz.
Isolados virais recuperados de CMSPs de pacientes envolvidos em estudos clínicos com o efavirenz e que mostraram evidência de falha terapêutica (rebote da carga viral) foram avaliados quanto à sensibilidade aos ITRNNs. Treze isolados anteriormente caracterizados como resistentes ao efavirenz foram também resistentes à nevirapina e à delavirdina. Observou-se que cinco desses isolados resistentes aos ITRNNs apresentavam substituição na posição K103N ou uma substituição valina-para isoleucina na posição 108 (V108I) na TR. Três dos isolados testados que não apresentaram resposta ao tratamento com o efavirenz permaneceram sensíveis ao efavirenz em cultura celular e também foram sensíveis à nevirapina e à delavirdina.
O potencial de resistência cruzada entre o efavirenz e os inibidores da protease é baixo por causa dos diferentes alvos enzimáticos envolvidos. O potencial de resistência cruzada entre o efavirenz e os ITRNs é baixo em função dos diferentes locais de ligação no alvo e mecanismos de ação.
Interação alimentar
Efavirenz pode ser ingerido com ou sem alimento.
A administração de Efavirenz com alimentos pode aumentar a exposição ao efavirenz e pode levar a um aumento na freqüência de efeitos adversos. Tomar Efavirenz com o estômago vazio, de preferência antes de se deitar, pode ser considerado.
Fonte
Fonte: Bula do Profissional do Medicamento Stocrin ® .